Deep learning enables rapid identification of potent DDR kinase inhibitors Nature Biotechnology 2019, 1038.
少し前の報告ですが、Insilicomedicineというアメリカのベンチャー企業から発表されたもので、GENTRL (GEnerative Tensorial Reinforcement Learning) というディープラーニング手法を用いて2ヶ月で10 nM程度のヒット化合物を取得したというものです。以前のエントリでもAIを用いた創薬例を紹介しましたが、ある程度相手が見えている場合においてはかなり実戦レベルの技術に仕上がってきているという印象です。
実際の流れですが、市販化合物から構築された1300万程度のopen virtual libraryであるZINCを出発点とし、既存のDDR1阻害化合物およびその他のキナーゼ阻害化合物を入力し強化学習を行うことで30,000構造を選抜しました。そこからアラート構造と反応性化合物を除去したのち、クラスタリングしてその代表化合物を選抜しました。さらに既知DDR1阻害化合物とタンパクのX線共結晶構造を使ってファーマコフォア解析を実施、40化合物がファーマコフォアに適合するものとして選ばれました。このうち特許面をクリアしていた39化合物から合成可能性の観点で6化合物を最終的に選抜しました。選抜された6化合物の合成と評価を実施したところ、4化合物にはDDR阻害活性があり、1化合物は10 nMとかなりの高活性でした。
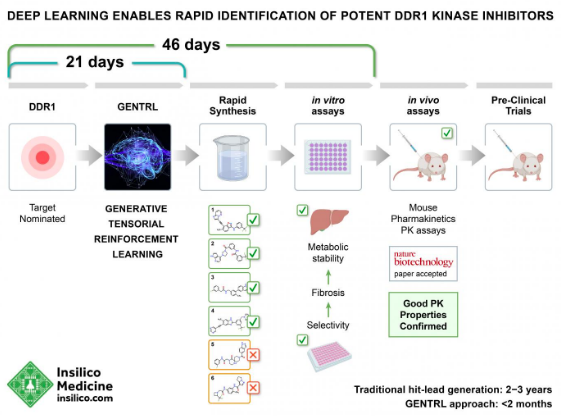
トータルで2ヶ月、化合物デザインはわずか1ヶ月でnMヒット、これはすごいです。DDR1は10 nM程度の既知阻害剤があるようなのですが、得られたヒットは骨格も既知阻害剤からはかなり離れているので、単純なMe tooの合成展開でここまで辿り着こうとすると1年くらいは必要でしょう。しかも6化合物から4化合物を当てるのは半端じゃないです。
じゃあ低分子創薬はAIで、となるかと言うとまだまだ、というかかなり突っ込みどころもあります。
まずDDR1はキナーゼなのですが、キナーゼは高活性化合物がかなり取得しやすいターゲットです。キナーゼにはリン酸化に用いるATPの結合サイトが必ずあるのですが、これは各キナーゼ間で共通性が高く、結合しやすい低分子の構造も広く理解されています。なので他のキナーゼの阻害剤からDDR1の高活性化合物が得られる可能性はかなり高いです。しかもDDR1は結晶構造も報告されているようなので、(活性向上の観点で言えば)かなり創薬しやすい標的となります。
そしてそれに関連する項目として、一番に思うのが「キナーゼの難しさは活性向上でなく選択性」ということです。キナーゼ阻害薬研究においては、ATP結合サイトを狙っている限り他のキナーゼとの選択性が毒性の観点から常に課題となります。ここがキナーゼ研究の肝であり、ヒットを作るところは多少の時間は必要であれ、クリティカルパスではないです。
おそらくキナーゼ研究のナレッジ蓄積がある製薬会社であれば、結晶構造のポケットの形を見ながら自社のキナーゼライブラリから筋の良い化合物を選抜するということも可能であると思います。であれば、最終的なゴールまでの距離はこちらの方がかなり近いと思います。
ということで、前回のエントリと同じく今回の報告にも「明確な答えがあるところで効率よくできる」という印象を持ちました。あえて対立構造はないのですが、メドケムの視点から競合として考えると、「Pharmacologyを含めPJを総合的に考えられるメドケム」にとってはまだ脅威でないが、「合成展開が仕事のメドケム」はAIに置き換わられる日が近づいているように思います。メドケムのチームは「リーダー + AI&委託合成」が主流になっちゃいそうなので、自分も強みを磨いて生き残れるリーダーになれるよう頑張ります。。。





コメントを残す